Executive Summary
Title of trial
Integrated Therapies for Alcohol use in Alcohol-associated Liver Disease (ITAALD)
Name of Active Ingredient
- F-652 (IL-22)
- Prednisone
- Acamprosate
Pharmacological Class of the Drugs
- F-652 (IL-22) is a fusion protein of human IL-22 with IgG2 fragment, and has anti-inflammatory effects
- Prednisone is an adrenal glucocorticoid with anti-inflammatory effects
- Acamprosate is a propane-1 sulfonic acid with anti-ethanol dependency effects
Indication
Severe alcohol-associated hepatitis (sAH), steroid-eligible
Study Type
Phase 2B, multicenter, sequentially randomized controlled trial for integrated treatments.
Investigational Sites
Indiana University, Mayo Clinic, Virginia Commonwealth University, Cleveland Clinic Foundation, University of Louisville, and University of Texas Southwestern Medical Center
Planned number of Participants
A total of 216 participants with sAH will be enrolled sequentially randomized into four treatment groups
Objectives:
- To determine whether interventions directed to treat AUD integrated with sAH therapies will improve a composite endpoint of alcohol and liver-related events at 6 months compared to usual care for AUD (primary endpoint).
- To compare 90-day survival in patients receiving F-652 with those receiving up to 28 days of prednisone using the Day-7 Lille score as a stopping rule (secondary endpoint).
- To compare one-year overall survival in patients receiving either IL-22 or prednisone with or without acamprosate (secondary endpoint).
Trial design and conduct
We will conduct a prospective, multicenter, sequentially randomized trial in 216 patients with sAH using a sequentially randomized design for proof of concept. The trial will assess whether integrated treatment of sAH and alcohol use disorder (AUD) reduces alcohol- and liver-related events and mortality.
The trial design resembles a 2X2 factorial study, but the AUD treatment assignment [acamprosate + motivational interviewing (MI) + motivational enhancement therapy (MET)] versus usual care (UC), defined as a brief intervention with advice not to drink alcohol-containing beverages and referral to a 12-step program, is done on Day 7 after the start of the sAH treatment. Only survivors of the first 7 days will be randomized to receive the AUD intervention or UC.
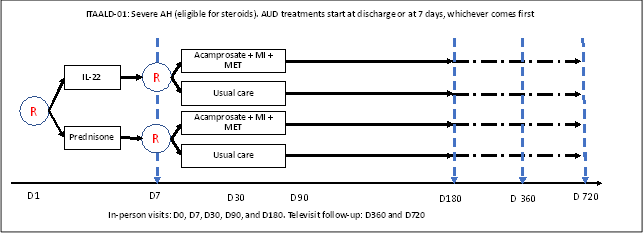
Figure 1. Diagram of trial design
Abbreviations: R: randomization, AUD: alcohol use disorder, MI: motivational interviewing, MET: motivational enhancement therapy.
The study will be conducted at six clinical sites in the United States selected by the National Institute of Alcoholic Abuse and Alcoholism (NIAAA) and supported by a Data Coordinating Center at Indiana University (DCC). The DCC will also manage the biorepository.
The study will be conducted according to Good Clinical Practice (GCP) and in compliance with local, state, and federal regulatory requirements. Adverse events during the trial will be identified, recorded, assessed for causality, and reported in accordance with FDA guidance. In addition to general assessment, we will specifically focus on (1) rates and types of infection as well as their severity, (2) potential development of drug-induced liver injury, (3) injection site reactions, and (4) hematological adverse events.
This study will be approved by an appropriately convened single IRB, Advarra, and will be monitored by an NIAAA-appointed Data and Safety Monitoring Board (DSMB). We will conduct this study under an Investigational New Drug (IND) application from the United States Food and Drug Administration (FDA).
It is anticipated that a centrally located investigational pharmacy at Indiana University will dispense the study medications to all participating sites under close coordination from the DCC. The study will provide the participants with F-652, prednisone, matching placebos, and acamprosate.
Duration of Trial by Phase
- Treatment phase: up to 6 months
- Follow-up phase: up to two years
Interventions to be tested
F-652 vs. prednisone as treatment for sAH.
AUD interventions (acamprosate + MI + MET) versus usual care for AUD treatment.
Group assignment:
We will sequentially randomize the study participants into four treatment groups.
On day 1, participants will be randomized to receive either F-652 or prednisone at a 1-to-1 ratio. Those randomized to receive F-652 will also receive a prednisone placebo; those randomized to receive prednisone will receive the F-652 placebo. Prednisone or prednisone placebo will be stopped if the Day-7 Lille score is >0.45.
On day 7, survivors will be randomized to receive either the AUD intervention (acamprosate + MI + MET) or usual care at a 1-to-1 ratio.
Block randomization will be used to ensure balanced group sizes in randomization.
So, there will be four treatment combinations:
- ARM 1: F-652 on days 1 and 7 and matching placebos for prednisone for 28 days and acamprosate for 6 months. MI will be delivered during the hospitalization; MET sessions will be delivered in the first 3 months.
- ARM 2: F-652 on days 1 and 7 and matching placebo for prednisone for 28 days and usual care for AUD.
- ARM 3: Prednisone for 28 days and matching placebos for F-652 on days 1 and 7 and acamprosate for 6 months. MI will be delivered during the hospitalization; MET sessions will be delivered in the first 3 months.
- ARM 4: Prednisone for 28 days and matching placebo for F-652 on days 1 and 7 and usual care for AUD.